RNA molecules are demonstrating a surprising breadth of biological functions. The repertoire of known non-protein-coding RNAs (ncRNAs) has grown extremely rapidly. Through all those discoveries, a new understanding of gene expression regulation is emerging.
Herein, we take an important step to help better understand the cellular roles of RNA by predicting their sub-cellular localization. The advent of widely available frameworks for deep learning (e.g., Scikit-Learn and TensorFlow) as well the recent release of RNALocate, a database for RNA subcellular localization, are making this project possible and timely.
Specifically, the project aims to answer the following research question: Can the subcellular localization of RNA molecules be predicted in silico from sequence information only?
Artificial Intelligence has become a hot topic again. Through the groundbreaking work of Jeff Hinton (University of Toronto) and Yoshua Bengio (Universit de Montral), Canada is now playing a leading role in machine learning. This work has found applications in research, but also in the industry. So much so that all the leading high-tech companies (Google, Facebook, Microsoft, Thales, etc.) now have research laboratories in Canada.
Throughout this project, the student will learn skills that are in high demand by the industry, locally, nationally and worldwide. He will develop skills preparing data for machine learning experiments. He will carefully design the research protocol to validate the results. He will evaluate the ability of different frameworks and deep neural network architectures to predict the localization of RNA molecules from sequence information only.
The project will consist of the following steps: 1) Review the literature on deep learning in bioinformatics (2 weeks). Data preparation (3 weeks): Writing parsers to extract the sequence information from the RNALocate database. Prepare the data to remove redundancy. Design the cross-validation protocol (2 weeks). Carry out the experiment (3 weeks). Analyze the results (2 weeks): Writing the abstract. Creating the poster presenting this work.
- Angermueller, C., Prnamaa, T., Parts, L. & Stegle, O. Deep learning for computational biology. Mol Syst Biol 12, (2016).
- Jurtz, V. I. et al. An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 33, 36853690 (2017).
- Min, S., Lee, B. & Yoon, S. Deep learning in bioinformatics. Brief Bioinform 18, 851869 (2017).
- Zhang, T. et al. RNALocate: a resource for RNA subcellular localizations. Nucleic Acids Res 45, D135D138 (2017).
This project is similar in nature to the one above except for the data set. A machine learning approach will be used for the analysis of RNA sequences packaged in exosomes, which cell-derived vesicules.
- Li, S., Li, Y., Chen, B., Zhao, J., Yu, S., Tang, Y., et al. (2018). exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Research 46(D1), D106D112. http://doi.org/10.1093/nar/gkx891.
See Jurtz, V. I. et al. An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 33, 36853690 (2017).
- The algorithm explores the space of regular expressions and identifies expressions that are enriched in the test set compared to the control set. At the end of the process, the regular expressions are converted into Position Specific Weight Matrices (PSWMs) in an ad hoc manner, the nucleotides are equiprobable. The project would consist of motifying the code that produces the PSWMs and use the actual matched sequences. The code is written in Python. It suffices to use matching groups in the expressions to collect sequence information and create PSWMs based on actual nucleotides frequencies. The hypothesis to be tested would that it would improve the E-value.
- Currently, the positions of the motif are considered to be independant one from another. Recent research has shown that it could be advantageous to consider dependencies. Regular expressions in Python have lookbehind and lookahead extensions that could be used to implement dependencies. This could be done in various ways: including allowing the evolutionary algorithm to select where these dependencies should be added, or having depencies for all the positions. The project would consist of testing the following hypothesis: expressions with dependencies have better P-values.
- MotifGP implements a multi-objective optimization. It would be interesting to explore adding new objectives: normalized complexity, entropy, GROMER, etc. See “C. K. Kibet and P. Machanick, Transcription factor motif quality assessment requires systematic comparative analysis, F1000Research, vol. 4, p. 1429, Mar. 2016.” for ideas.
Seed is a computer program that takes as input a set of unaligned RNA sequences and produces a set of secondary structure motifs. Suffix arrays are used enumerate complementary regions, possibly containing interior loops, as well for matching RNA secondary structure expressions.
Seed has several criteria to rank the motifs that it produces: minimum description length, information theory, and free energy.
In order to enable one of our future research directions, it would be interesting to see if deep learning can be used to rank the motifs produced by Seed.
The project consists of: developping a deep neural network architecture, using information from Rfam to train the model, using a rigourous cross-validation strategy, compare the accuracy of deep learning to classify motifs against the existing criteria that used by Seed.
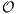 (n2) time (and
space) for comparing two sequences of length (n) each, which limits their application to relatively
short segements. An alternative approach consists of comparing k-word frequencies of the two
sequences. Implement and benchmark an alignment-free sequence comparison method. (Vinga et al.
Comparative evaluation of word composition distances for the recognition of SCOP relationships.
Bioinformatics (2004) vol. 20 (2) pp. 206-15; Sims et al. Alignment-free genome comparison with feature
frequency profiles (FFP) and optimal resolutions. Proc Natl Acad Sci USA (2009) vol. 106 (8) pp.
2677-82)
(n2) time (and
space) for comparing two sequences of length (n) each, which limits their application to relatively
short segements. An alternative approach consists of comparing k-word frequencies of the two
sequences. Implement and benchmark an alignment-free sequence comparison method. (Vinga et al.
Comparative evaluation of word composition distances for the recognition of SCOP relationships.
Bioinformatics (2004) vol. 20 (2) pp. 206-15; Sims et al. Alignment-free genome comparison with feature
frequency profiles (FFP) and optimal resolutions. Proc Natl Acad Sci USA (2009) vol. 106 (8) pp.
2677-82)
www.khronos.org/opencl, www.macresearch.org/files/opencl
The BioCatalogue (www.biocatalogue.org) is a curated repertoire of Web services.
- M. N. Davies and D. R. Flower. (2007) Harnessing bioinformatics to discover new vaccines. Drug Discov Today, 12(9-10):389–95.
- J. A. Greenbaum, et al (2007) Towards a consensus on datasets and evaluation metrics for developing b-cell epitope prediction tools. J Mol Recognit, 20(2):75–82.
(km) time/space, where m is the size of the longest of the two input strings. This project
requires developing a Java implementation of such algorithm using Daniela’s suffix tree library, see Gusfield p.
268 for a description. (A)