Essential Cell Biology (Part 1)
CSI 5180 - Machine Learning for Bioinformatics
Version: Jan 13, 2025 12:00
Preamble
Quote of the Day
Summary
In this lecture, we will explore the cell, including the different types of cells, their organisation, and composition. We will also introduce concepts from molecular evolution. Additionally, we will discuss the macromolecules that make up the cell and their basic structures. Throughout the lecture, we will emphasize the relevance of these concepts to machine learning and bioinformatics.
General objective
- Describe the organisation of the cell and its macromolecules
Learning Outcomes
- Describe the organization, composition, and types of cells, differentiating between prokaryotic and eukaryotic structures.
- Explain the structural and functional significance of cellular macromolecules (DNA, RNA, and proteins).
- Illustrate the hierarchical structure of macromolecules, identifying primary, secondary, tertiary, and quaternary levels.
- Summarize the phylogenetic relationships among organisms and the relevance of molecular evolution in bioinformatics.
- Interpret bioinformatics challenges related to sequence alignment, gene prediction, and structural inference in cellular biology.
- Discuss the relevance of three-dimensional genome organization in understanding regulatory mechanisms.
- Analyze the historical and contemporary integration of computational and biological perspectives in molecular biology.
Machine learning & Dangerous Pathogenic Sequences

Symbiosis
- Symbiotic interactions encompass mutually beneficial relationships (mutualism) or harmful interactions where one organism, the parasite, adversely affects the other (parasitism). Key objectives include:
- Promoting beneficial interactions;
- Preventing detrimental interactions.
- Microbiome Host Trait Prediction: Advances in this area have significant implications for fields such as medicine and agriculture, among others.
Did You Know?
Tip
With a uOttawa IP address, you can access 300,000 books for free from Springer Nature Link. This includes full downloads in PDF or EPUB formats.
Further Reading

- A Computer Scientist’s Guide to Cell Biology (Cohen and Cohen 2024). Provides an “introduction for computer scientists to the exciting revolution underway in molecular biology.”
- Full downloads in PDF or EPUB format from Springer Nature Link.
- In this second edition, the authors introduce concepts such as COVID-19 and CRISPR/Cas9.
- 117 pages.
Further Reading

- Molecular Biology: Not Only for Bioinformaticians (Widła 2013). “Provides beginners with an introduction to molecular biology”
- Full download in PDF format from Springer Nature Link.
- 153 pages.
The Cell
Cell Structure
Cells: Fundamental Units of Life
Cells are categorized based on the presence or absence of a nucleus:
Prokaryotes: These cells or organisms lack a membrane-bound, structurally distinct nucleus and other sub-cellular compartments. Bacteria exemplify prokaryotic life forms.
Eukaryotes: These cells or organisms possess a membrane-bound, structurally distinct nucleus along with well-developed sub-cellular compartments. Eukaryotes comprise all organisms except viruses, bacteria, and cyanobacteria (blue-green algae).
Cells: Fundamental Units of Life
Eukaryotic cells typically exhibit larger dimensions than their prokaryotic counterparts.
In eukaryotic cells, genetic material (DNA) is organised and condensed more intricately than in prokaryotic cells.
Prokaryotic vs Eukaryotic Cell
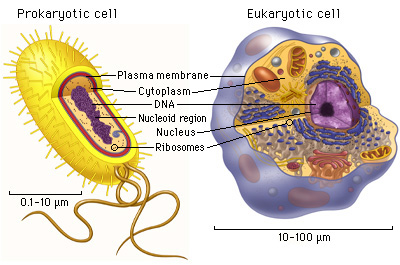
Organisation of an Eukaryotic Cell

Organelle Genomes
- Organelles are distinct cellular structures characterized by specialized functions.
- Mitochondria function as the cell’s powerhouses, facilitating energy production.
- These organelles possess their own DNA, encompassing a limited set of genes referred to as mitochondrial or extrachromosomal genes.
- The endosymbiotic theory, notably advanced by Lynn Margulis, postulates that certain organelles originated from engulfed prokaryotes.
- Mitochondrial genes are exclusively maternally inherited.
Bioinformaticist’s Perspective
- Gene Organisation: Genome structures vary significantly between cell types, necessitating tailored gene-finding algorithms.
- Complexity of Eukaryotic Cells: The intricate nature of eukaryotic cells introduces complex challenges, such as the protein sub-cellular localization issue.
- Sequence Assembly Considerations: During sequence assembly, it is crucial to account for potential contamination sources, including mitochondrial DNA, nuclear DNA, and bacterial DNA.
Resources
Kingdoms of Life
(3) Kingdoms of Life
Prokarya: Organisms classified as prokaryotes lack a defined nucleus. Exemplary species include Cyanobacteria (blue-green algae) and Escherichia coli (common bacteria).
Eukarya: Eukaryotic organisms possess cells with a well-defined nucleus. Examples include Trypanosoma brucei (a unicellular organism known for causing sleeping sickness) and Homo sapiens (a multicellular organism).
Archaea: Although archaea lack a nuclear membrane similar to prokaryotes, their transcription and translation mechanisms are more akin to those found in eukaryotes.
(3) Kingdoms of Life: Archaea
Methanococcus jannaschii is a methanogenic archaeon, notable for being the first archaebacterium to have its entire genome sequenced in 1996. Discovered in 1982, this organism inhabits the extreme environment of a white smoker vent at the Pacific Ocean’s seabed, located 2,600 meters deep. It thrives in temperatures ranging from 48°C to 94°C, with an optimal growth temperature of 85°C. Its genome comprises 1.66 megabases and includes 1,738 genes. Remarkably, 56% of these genes show no homology to those found in eukaryotic or prokaryotic organisms. Additionally, M. jannaschii possesses a single type of DNA polymerase, in contrast to the multiple types typically present in other genomes.
Phylogenetic Tree

Phylogenetic Tree
“The objectives of phylogenetic studies are (1) to reconstruct the genealogical ties between organisms and (2) to estimate the time of divergence between organisms since they last shared a common ancestor.”
“A phylogenetic tree is a graph composed of nodes and branches, in which only one branch connects any two adjacent nodes.”
“The nodes represents the taxonomic units, and the branches define the relationships among the units in terms of descent and ancestry.”
“The branch length usually represents the number of changes that have occurred in that branch.” (or some amount of time)
Bioinformaticist’s Perspective
- Benchmarking and Cross-Validation: Impact on the assumption of independence.
- Molecular Sequence Alignment: are the sequences evolutionary related?
- Large Phylogeny Problem: Reconstructing phylogenetic trees from molecular sequence data.
- Small Phylogeny Problem: Reconstructing ancestral molecular sequences.
Theodosius Dobzhansky
- Nothing in Biology Makes Sense Except in the Light of Evolution.
What About Viruses?
- Viruses are infectious agents that invade the cells of living organisms.
- They cannot self-replicate and therefore must exploit the host cell’s machinery for reproduction.
- Structurally, viruses are composed of nucleic acids encased in protein shells.
- Their genomes are compact, primarily encoding proteins that form the protective capsid.
- Viral genomes may consist of either DNA or RNA.
- Some RNA viruses (termed retroviruses) possess an enzyme called reverse transcriptase, which transcribes their RNA genome into DNA, facilitating integration into the host genome.
- Bacteriophages, or phages, specifically target bacterial cells.
- In contrast, viroids lack a capsid and are composed solely of single-stranded RNA.
BNT162b2 mRNA vaccine
- Reverse Engineering the source code of the BioNTech/Pfizer SARS-CoV-2 Vaccine by bert hubert, an entrepreneur & software developer.
Composition of the Cell
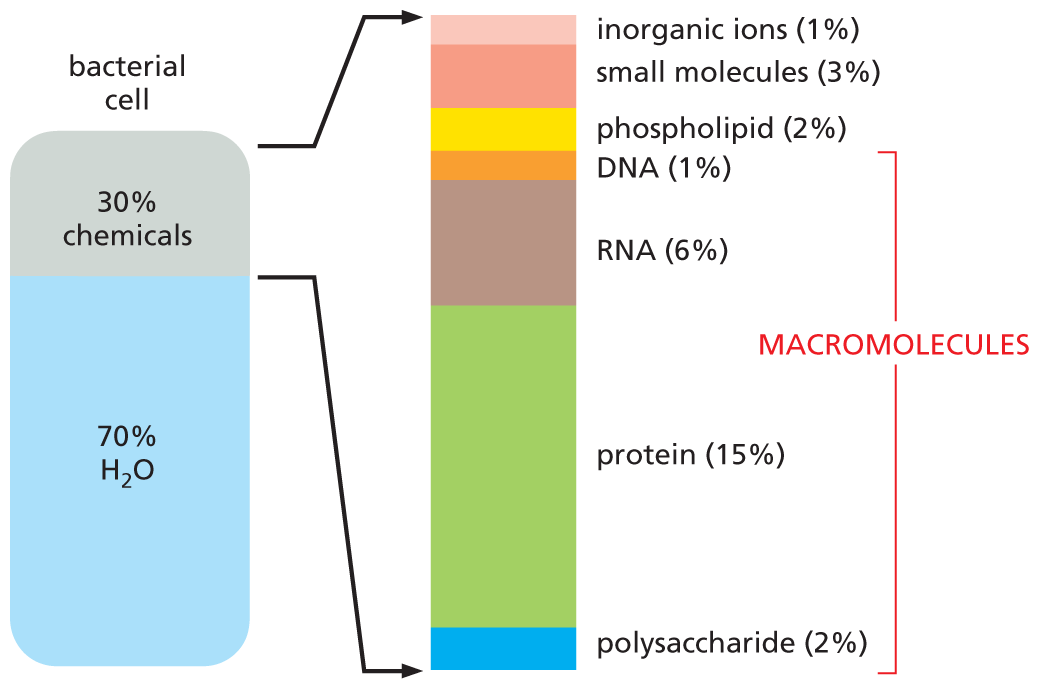
Macromolecules
DNA, RNA and Protein
- Bioinformatics is mainly concerned with three classes of molecules:
- DNA (deoxyribonucleic acid), RNA (ribonucleic acid) and proteins — collectively called macromolecules or biomolecules.
Polymers
- All three macromolecule classes are polymers, composed of sequentially linked monomers that form unbranched linear structures.
Monomers
Monomers comprise two distinct components: a common segment that forms the molecular backbone shared by all monomers, and a unique segment that determines the monomer’s identity and properties.
ASCII (Unicode) representation of a polymer:
[ ]-[ ]-[ ]-[ ]-[ ]- ... -[ ]-[ ]
| | | | | | |
* @ * # + + @Structure
We can categorize the structural hierarchy into four distinct levels of abstraction: primary, secondary, tertiary, and quaternary structures.
1, 2, 3
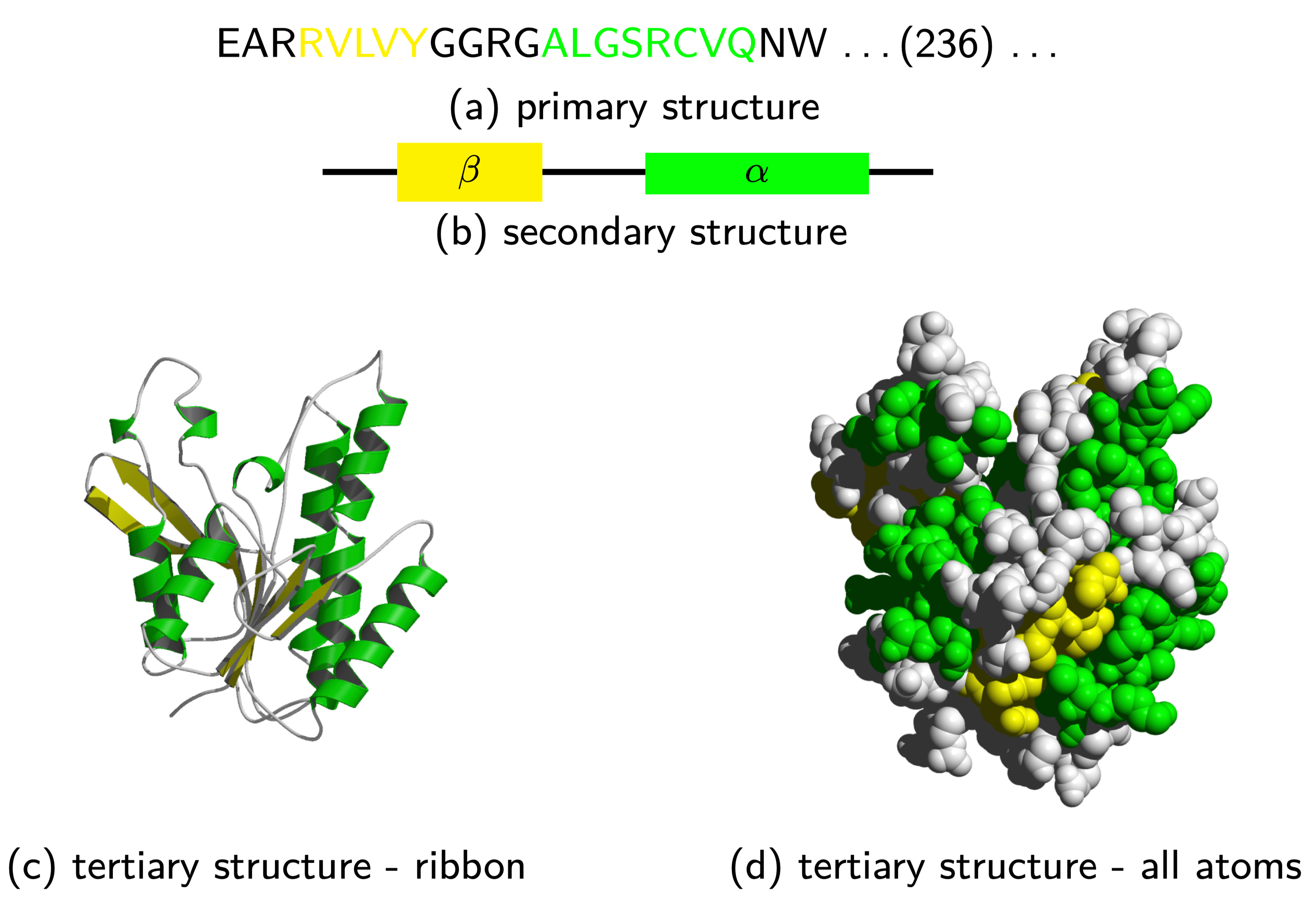
Bioinformaticist’s Perspective
- Numerous computational challenges center around the primary sequence, including sequence assembly, alignment, phylogenetic tree construction, gene prediction, and motif discovery.
- Predicting secondary, tertiary, and quaternary structures (docking), constitutes distinct problem sets.
- These abstractions facilitate the development of efficient algorithms, underscoring the importance of comprehending their implications.
DNA, RNA and Protein
The primary structure or sequence is an ordered list of characters, from a given alphabet, written contiguously from left to right.
DNA (deoxyribonucleic acid): 4 letters alphabet,
\(\Sigma = \{A,C,G,T\}\)RNA (ribonucleic acid): 4 letters alphabet,
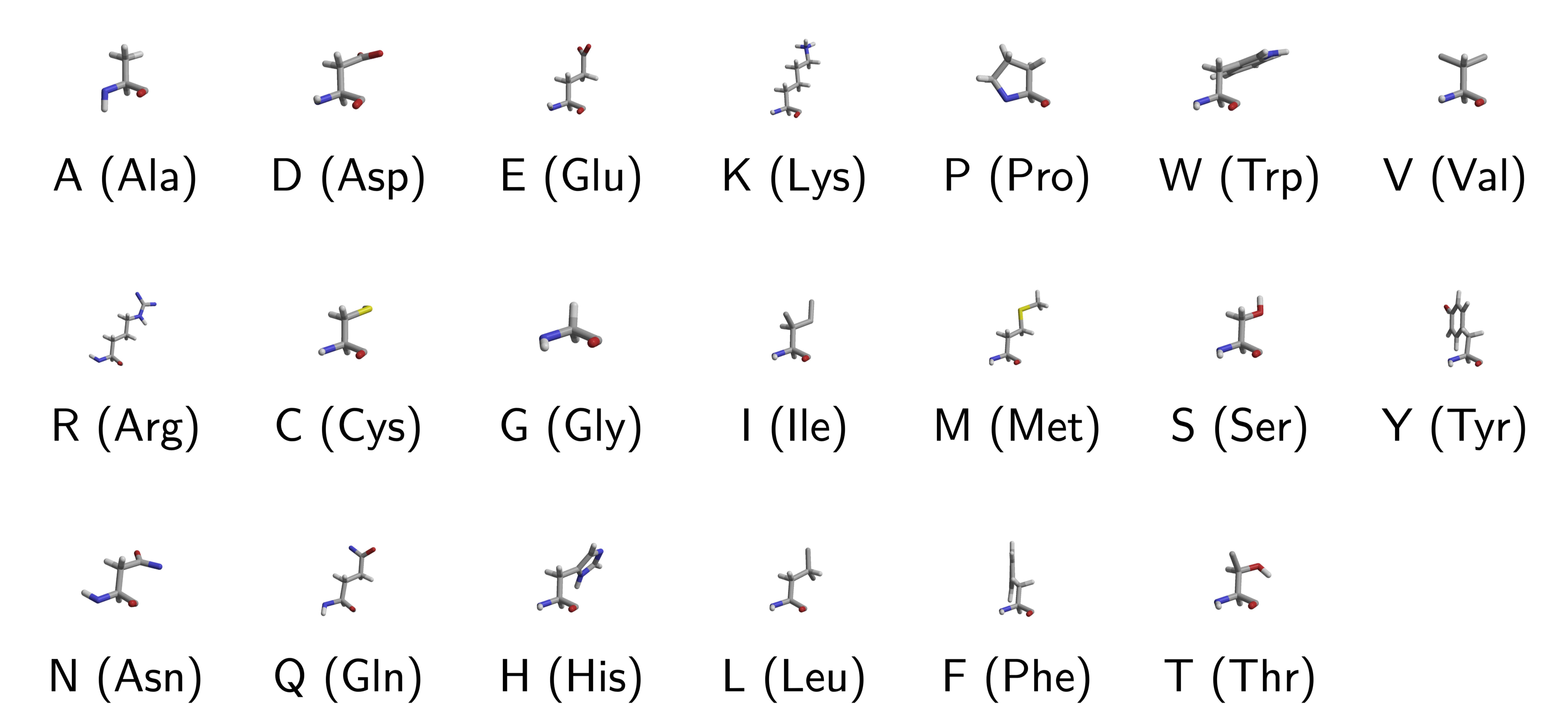
\(\Sigma = \{A,C,G,U\}\)Proteins: 20 letters alphabet,
\(\Sigma = \{A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y\}\)
Examples
In the case of nucleic acids (DNA and RNA), the building blocks are called nucleotides, whilst in the case of proteins they are called amino acids.
Examples of DNA, RNA and protein sequences in FASTA format.
> Chimpanzee Chromosome 1; A DNA sequence (size = 245,522,847 nt)
TAACCCTAACCCTAACCCTAACCCTAACC ... TCTCATGACAGTGAGTGAGTTCTCATGATC> A01592; An RNA sequence (coding Beta Globin gene) (size = 441 nt)
AUGGUGCACCUGACUCCUGAGGAGAAGUCUGC ... GCAAGGUGAACGUGGAUGAAGUUGGUGGUGSequence File Formats
FASTA: This format is the most prevalent for representing the primary structure of proteins and nucleic acids. It consists of a header line starting with ‘>’, followed by lines of sequence data. It’s simple and widely supported by bioinformatics tools.
FASTQ: While primarily used for storing raw sequencing data, FASTQ files include sequence information and quality scores for each nucleotide, making it useful for primary sequence data from sequencing technologies.
GenBank: This format, maintained by the National Center for Biotechnology Information (NCBI), is used for nucleotide sequences and includes annotations. It captures the primary sequence along with additional information such as gene features and references.
EMBL: Similar to GenBank, the EMBL format stores nucleotide sequences with detailed annotations. It is used primarily in European databases.
Bioinformaticist’s point of view
- Exact string (sequence) comparison, approximate matching (\(k-\)mismatches), comparison under the edit-distance, significance of match, multi-way sequence comparison.
- Finding repeats, approximate repeats, finding interesting patterns.
- Secondary, tertiary and quaternary structure inference.
DNA
The Structure of DNA
DNA’s Building Blocks: ACGT
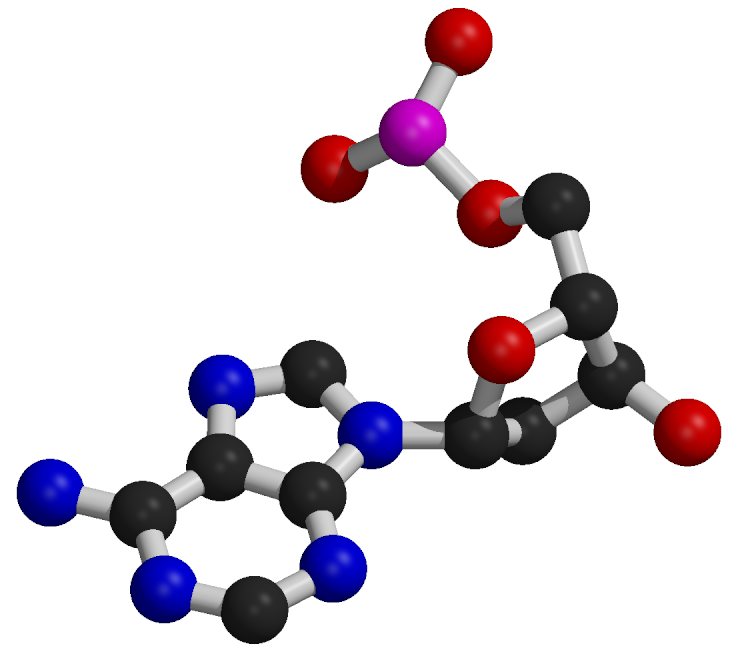
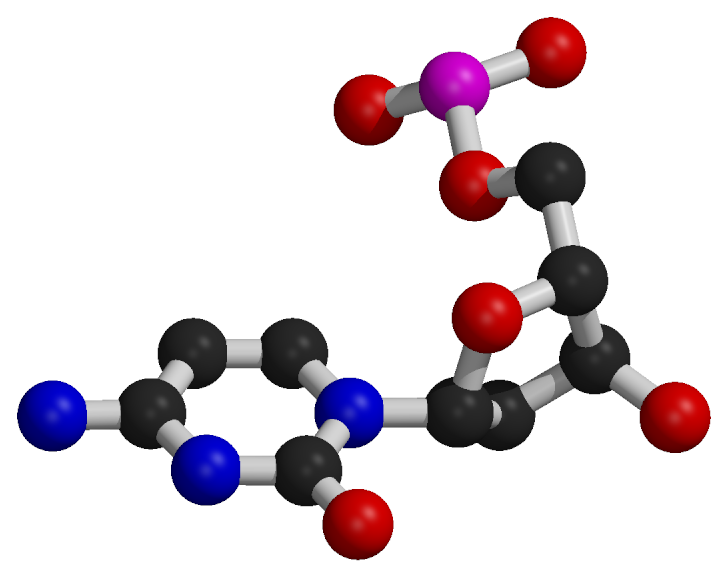
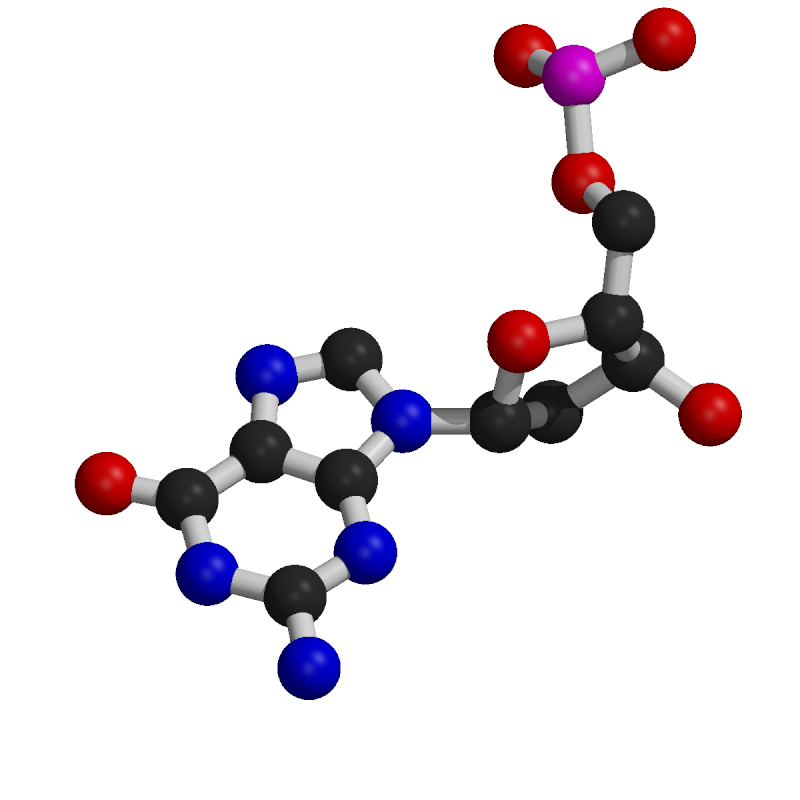
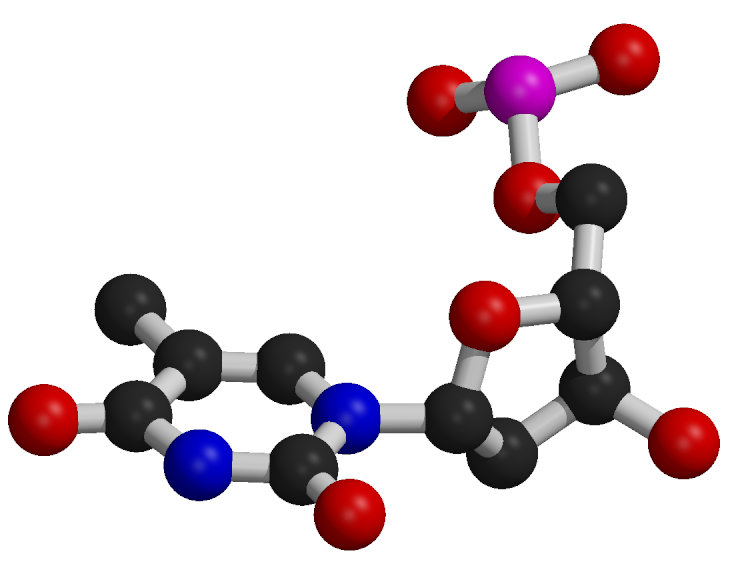
Nucleotide Structure
Nucleotides consist of a common component composed of a deoxyribose sugar (pentose) and a phosphate group.
The distinguishing feature of each nucleotide is the nitrogenous base.
Nitrogenous bases are categorized as purines, which are larger two-ring structures (adenine, A; guanine, G), and pyrimidines, which are smaller one-ring structures (cytosine, C; thymine, T).
In DNA, the nitrogenous bases include adenine (A), cytosine (C), guanine (G), and thymine (T).
In RNA, the bases are adenine (A), cytosine (C), guanine (G), and uracil (U), where uracil (U) replaces thymine (T).
Historical Notes
DNA was first identified by Johann Friedrich Miescher in 1869, who initially dismissed its role in heredity.
In 1953, James Watson and Francis Crick, with Crick passing on July 28, 2004, proposed the double-helical structure of DNA.
This structural elucidation is widely regarded as the pivotal biological discovery of the 20th century.
Their model provided a molecular basis for Chargaff’s rules, elucidating the equimolarity of adenine with thymine and guanine with cytosine.
Crucially, the model elucidated the mechanism by which DNA underpins heredity through replication.
Length Measurement
Length Measurement in Bases: The length of DNA or RNA molecules is commonly measured in bases. This is a standard unit of measurement for single-stranded nucleic acids. For example, a region 10 megabases long consists of 10 million bases.
Hybridization and Base Pairs: When nucleic acids hybridize, they form a double-stranded structure known as a duplex or double helix. In this context, the length is often measured in base pairs (bp) to reflect the paired nature of the strands. For instance, a region 10 megabase pairs (Mbp) long would consist of 10 million base pairs.
Nucleic Acid Fundamentals
- DNA and RNA Structure:
- DNA is deoxyribonucleic acid, named for the absence of an oxygen atom at the C2’ position of its sugar, deoxyribose. RNA is ribonucleic acid, with ribose sugar that has an oxygen atom at the C2’ position. This structural difference contributes to RNA’s functional versatility, such as its ability to act as a catalyst in some biological reactions.
- Nucleotide Bases:
- DNA uses thymine (T) as one of its four nitrogenous bases, while RNA uses uracil (U) instead of thymine.
DNA/RNA Building Blocks
DNA Orientation: The orientation of a DNA molecule is crucial, akin to word order in natural languages, impacting the interpretation and processing of genetic information.
5’ to 3’ Convention: DNA sequences are conventionally read from the 5’ to 3’ end. This directionality is essential for subsequent processes, which will be detailed later. Elements preceding the 5’ end are termed upstream, while those following the 3’ end are referred to as downstream, reflecting their relative positions in genetic signaling.
Strand

A, B, and Z form
DNA generally forms a right-handed double helix in the B form, which is the most common form of DNA in cells.
RNA typically forms an A form helix, which is also right-handed.
Z DNA is a known form of DNA that is a left-handed helix.
A DNA molecule is made of two complementary strands running in opposite directions, which refers to the antiparallel nature of the DNA double helix.
Watson-Crick (Canonical) Base Pairs
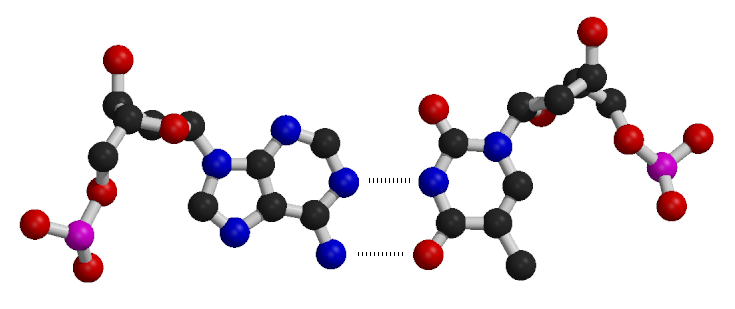
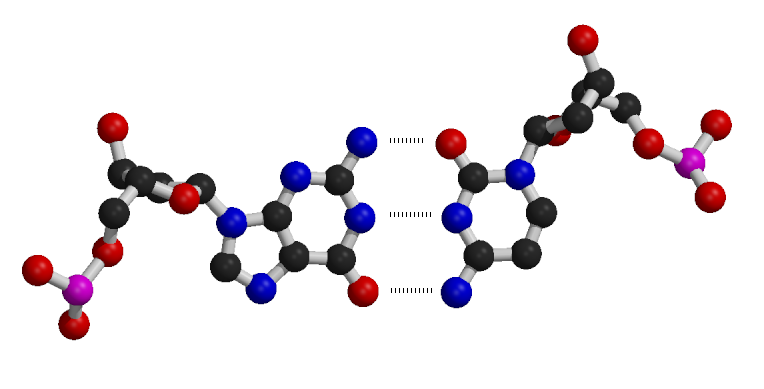
Watson-Crick (Canonical) Base Pairs
In DNA, nucleotide bases pair through hydrogen bonding according to specific rules:
- Adenine (A) pairs with Thymine (T)
- Guanine (G) pairs with Cytosine (C)
These pairing rules ensure that A:T and G:C pairs align backbone atoms similarly in three-dimensional space, maintaining the uniformity of the double helical structure due to their isosteric nature.
Linguistics and Bioinformatics (1/2)
The analogy between natural languages and molecular sequences is profound and extensive.
Since the 1950s, bioinformatics and linguistics have mutually influenced each other.
In bioinformatics, molecular sequences are analyzed using the edit distance (Levenshtein distance), a metric originally devised in computational linguistics.
Both fields employ tree structures to represent evolutionary relationships; bioinformaticians construct phylogenetic trees for molecular sequences, while linguists create analogous trees to depict the evolution of languages.
Linguistics and Bioinformatics (2/2)
Similarly, hidden Markov models (HMMs), a key machine learning technique, have been adapted from linguistic applications to bioinformatics. HMMs are used for speech recognition and gene prediction.
Currently, concepts such as embeddings, transformers, and large language models are increasingly applied in bioinformatics.
For example, EMS-2 is a transformer-based protein language model, trained on a dataset of 250 million protein sequences, illustrating the continuing exchange of methodologies between these domains.
Proteins
What is a Protein? (from PDB-101)
20 (Naturally Occuring) Amino Acids
Genome
Chromosome Structure
Chromatin
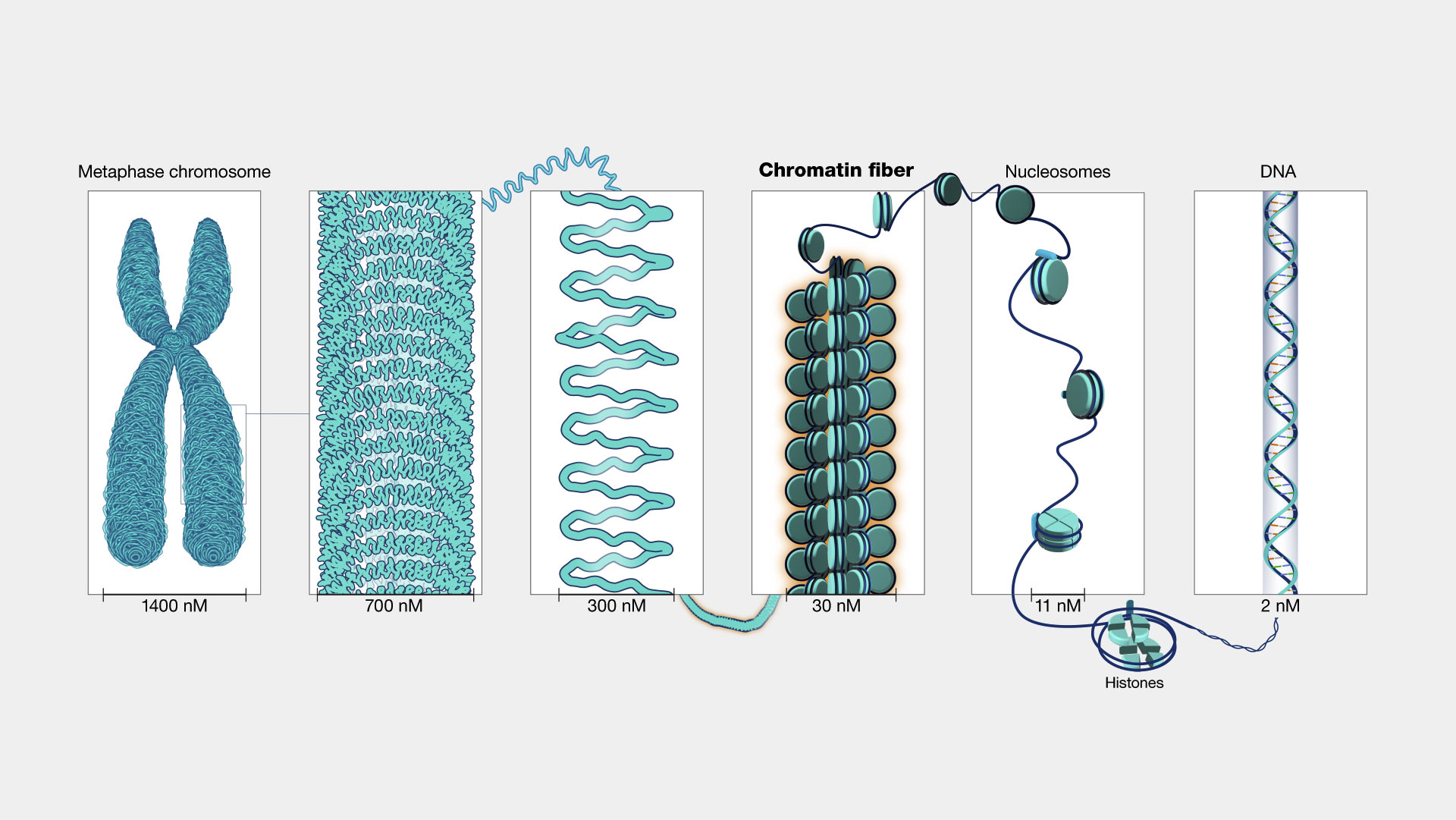
3D Organization of Our Genome (New)
Bioinformaticist’s Perspective
- Predicting histone binding sites based solely on DNA sequence data.
- Utilizing histone locations to infer gene positions and regulatory element sites.
- Exploring the three-dimensional genome organization, a current focal point in research.
Prologue
Summary
- Cellular life forms are categorized into two types: prokaryotic and eukaryotic.
- Eukaryotic cells possess organelles, with certain organelles, such as mitochondria, containing their own DNA.
- The three domains of life are Prokarya, Eukarya, and Archaea.
- A phylogeny delineates the evolutionary relationships among organisms and their divergence times.
- The primary macromolecules are DNA, RNA, and proteins.
- Macromolecules are linear, unbranched polymers, wherein each monomer comprises a common backbone and a distinct, specific component, akin to nodes in a linked chain.
Building the Paper Model of DNA
Next Lecture
- Essential Cell Biology (Part 2)
References
Marcel Turcotte
School of Electrical Engineering and Computer Science (EECS)
University of Ottawa