Essential Bioinformatics
CSI 5180 - Machine Learning for Bioinformatics
Version: Feb 10, 2025 09:12
Preamble
Quote of the Day
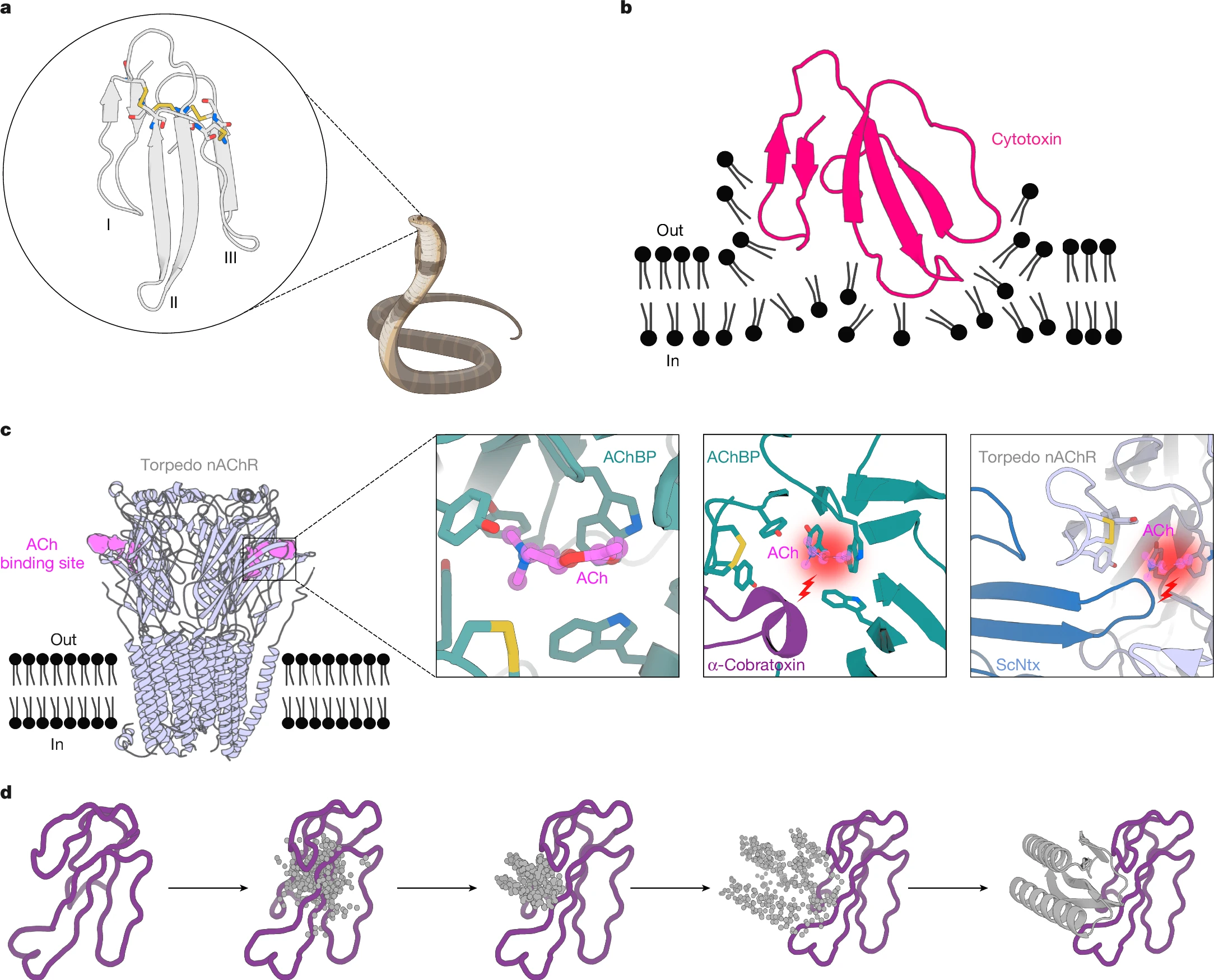
The AI-designed proteins could form the basis of a new generation of therapies for snakebites — which kill an estimated 100,000 people each year and are still treated much as they were a century ago.
Preamble
Summary
The lecture gives an overview of the available resources that are essential for bioinformatics projects. This includes the main databases, software applications, programming languages and computing environments. We also emphasize the skills that are essential to produce robust and reproducible results.
General objective:
- Summarize the essential resources for conducting a bioinformatics project
Remarks
You are advised not to hastily install all applications discussed today, as our goal is to review best practices. Only a specific subset of these tools will be necessary for your assignments and projects. Detailed instructions regarding the required tools will be provided for each task.
Nonetheless, proficiency in Jupyter Notebooks and Google Colab is recommended due to their anticipated utility in your coursework.
Learning objectives
- Describe the best practices for handling large bioinformatics projects
- Introduce essential tools
- Present the major repositories and file formats, along with the command line and REST API access
Python
For those needing a refresher, the official tutorial on Python.org is a good place to start.
Simultaneously enhance your skills by creating a Jupyter Notebook that incorporates examples and notes from the tutorial.
Other resources include:
Jupyter Notebooks
Deep Learning Genomics Primer

Jupyter Notebooks
A notebook is a shareable document that combines computer code, plain language descriptions, data, rich visualizations like 3D models, charts, graphs and figures, and interactive controls. A notebook, along with an editor (like JupyterLab), provides a fast interactive environment for prototyping and explaining code, exploring and visualizing data, and sharing ideas with others.
Quick Start
![]()
Running Jupyter on Your Computer
Assuming the notebook is in the current directory, execute the following command from the terminal.
Similarly, to create a new notebook from scratch,
Why?
Ease of Use: The interface is intuitive and conducive to exploratory analysis.
Visualization: The capability to embed rich, interactive visualizations directly within the notebook enhances its utility for data analysis and presentation.
Reproducibility: Jupyter Notebooks have become the de facto standard in many domains for demonstrating code functionality and ensuring reproducibility. Suggested reading: Samuel and Mietchen (2024).
How?
- Google Colab
- Local installation
- In your browser
- Notebook or JupyterLab
- Visual Studio Code
- In your browser
- More options, including JupyterHub (a multi-user version)
Google Colab
We will employ numerous libraries, such as NumPy, Pandas, Scikit-learn, Keras, TensorFlow or PyTorch, Matplotlib, and Seaborn, among others.
The installation process for these libraries involves dependencies that total around 100 additional packages, potentially causing conflicts with existing projects.
Installing Jupyter (1/2)
These instructions use pip, the recommended installation tool for Python.
The initial step is to verify that you have a functioning Python installation with pip installed.
Installing Jupyter (2/2)
Installing JupyterLab with pip:
Once installed, run JupyterLab with:
Sample Jupyter Notebooks
Missing libraries
Launching 02_interactive_3d_viewert in Colab.
![]()
Version Control (GitHub)
By default, Jupyter Notebooks store the outputs of code cells, including media objects.
Jupyter Notebooks are JSON documents, and images within them are encoded in PNG base64 format.
This encoding can lead to several issues when using version control systems, such as GitHub.
- Large File Sizes: Jupyter Notebooks can become quite large due to embedded images and outputs, leading to prolonged upload times and potential storage constraints.
- Incompatibility with Text-Based Version Control: GitHub is optimized for text-based files, and the inclusion of binary data, such as images, complicates the process of tracking changes and resolving conflicts. Traditional diff and merge operations are not well-suited for handling these binary formats.
Version Control (GitHub) - solutions
- In JupyterLab or Notebook, Edit \(\rightarrow\) Clear Outputs of All Cells, then save.
Environment Management
Environment Management
Important
Do not attempt to install these tools unless you are confident in your technical skills. An incorrect installation could waste significant time or even render your environment unusable. There is nothing wrong with using pip or Google Colab for your coursework. You can develop these installation skills later without impacting your grades.
Package management
- Managing package dependencies can be complex.
- A package manager addresses these challenges.
- Different projects may require different versions of the same libraries.
- Package management tools, such as
conda, facilitate the creation of virtual environments tailored to specific projects.
- Package management tools, such as
Anaconda
Anaconda is a comprehensive package management platform for Python and R. It utilizes Conda to manage packages, dependencies, and environments.
Anaconda is advantageous as it comes pre-installed with over 250 popular packages, providing a robust starting point for users.
However, this extensive distribution results in a large file size, which can be a drawback.
Additionally, since Anaconda relies on
conda, it also inherits the limitations and issues associated withconda(see subsequent slides).
Using conda/anaconda/bioconda
$ conda create -n csi5180$ conda create -n csi5180 python=3.10$ conda install -n csi5180 keras$ conda activate csi5180$ conda install bwa$ conda update --all$ conda deactivate$ conda remove --name csi5180 --all
Miniconda
Miniconda is a minimal version of Anaconda that includes only conda, Python, their dependencies, and a small selection of essential packages.
Conda
Conda is an open-source package and environment management system for Python and R. It facilitates the installation and management of software packages and the creation of isolated virtual environments.
Dependency conflicts due to complex package interdependencies can force the user reinstall Anaconda/Conda.
Plague with large storage requirements and performance issues during package resolution.
Mamba
Mamba is a reimplementation of the conda package manager in C++.
- It is significantly faster than
conda. - It consumes fewer computational resources.
- It provides clearer and more informative error messages.
- It is fully compatible with
conda, making it a viable replacement.
Micromamba is a fully statically-linked, self-contained executable. Its empty base environment ensures that the base is never corrupted, eliminating the need for reinstallation.
Conda/Mamba
- https://conda.io
- Conda is a package, dependency, and environment management for any programming language (Python, R, Ruby, Lua, Scala, Java, and more)
- https://anaconda.org
- Anaconda is a package management service, primarily for Python and R, including hundreds of packages such as numpy, scipy, scikit-learn, keras, tensorflow
- https://bioconda.github.io
- Bioconda is a channel for the conda package manager specializing in bioinformatics software.
Computing Environment
UNIX
Both, Bioinformatics and Machine Learning, favor UNIX.
Quoting François Cholette (Deep Learning with Python):
- “You’ll need access to a UNIX machine; it’s possible to use Windows, too, but I don’t recommend it.”
Digital Research Alliance
- Béluga - 38,960 CPU cores and 688 GPU devices (PolyMTL)
- Cedar - 93,712 CPU cores and 36,864 GPU devices (SFU)
- Graham - 36,160 CPU cores and 320 GPU devices (Waterloo)
- Narval - 83,216 CPU cores and 636 GPU devices (PolyMTL)
- Nibi - 134,400 CPU cores and 288 GPU divices (Waterloo)
- Niagara - 80,960 CPU cores (UofT)
Access to UNIX
- Your laptop or workstation
- macOS is certified Unix operating system
- As primary or secondary OS (dual boot, USB key, etc.)
- In a virtual machine
(VMWare is free for EECS students, VirtualBox is also free) - Windows Subsystem for Linux Installation Guide for Windows learn.microsoft.com/en-us/windows/wsl/install
- Cloud
UNIX Key Concepts
Modularity
“This is the Unix philosophy: Write programs that do one thing and do it well. Write programs to work together. Write programs to handle text streams, because that is a universal interface.” — Doug McIlory
UNIX Key Concepts
The file system plays a central role.
/dev/null,/dev/random, /dev/zero
UNIX Key Concepts
The command line
UNIX Key Concepts
Shell (anatomy of a script, the magic line, and more)
- Redirection
- Pipe
- www.ks.uiuc.edu/Training/Tutorials/Reference/unixprimer.html
Resources
- The Missing Semester of Your CS Education
- Introduction to Unix on Bioinformatics Workbooks
Data
Major repositories
Annotated/assembled nucleotide sequence
- National Center for Biotechnology Information (NCBI)
- European Bioinformatics Institute (EBI)
- DNA Data Bank of Japan (DDBJ)
See also: International Nucleotide Sequence Database Collaboration (www.insdc.org)
Major repositories (continued)
- GenBank: annotated and identified DNA sequence information
- SRA (Short Read Archive): measurements from high throughput sequencing experiments
- UniProt (Universal Protein Resource): protein sequence data
- PDB (Protein Data Bank): 3D structural information of macromolecules
Other data sources?
- UCSC Genome Browser
- FlyBase (Drosophila [fruit fly]), WormBase (nematode), SGD: Saccharomyces Genome Database, TAIR (Arabidopsis), EcoCyc (Encyclopedia of E. coli Genes and Metabolic Pathways), etc.
- RNA-Central: meta-database
Nucleic Acids Research (NAR)

Each year, NAR, a high-impact journal, publishes its “database issue”:
Major File Formats (biostar)
- Data that captures prior knowledge (aka reference: FASTA, GFF, BED)
- Experimentally obtained data (aka sequencing reads: FASTQ)
- Data generated by the analysis (aka results: BAM, VCF, formats from point 1 above, and many nonstandard formats)
Entrez Direct
GENBANK
LOCUS NM_000020 4177 bp mRNA linear PRI 16-SEP-2019
DEFINITION Homo sapiens activin A receptor like type 1 (ACVRL1), transcript
variant 1, mRNA.
ACCESSION NM_000020
VERSION NM_000020.3
KEYWORDS RefSeq; RefSeq Select.
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
REFERENCE 1 (bases 1 to 4177)
AUTHORS Leng H, Zhang Q and Shi L.
TITLE [Gene diagnosis and treatment of hereditary hemorrhagic
(...)GENBANK (continued)
(...)
FEATURES Location/Qualifiers
source 1..4177
/organism="Homo sapiens"
/mol_type="mRNA"
/db_xref="taxon:9606"
/chromosome="12"
/map="12q13.13"
gene 1..4177
/gene="ACVRL1"
/gene_synonym="ACVRLK1; ALK-1; ALK1; HHT; HHT2; ORW2;
SKR3; TSR-I"
/note="activin A receptor like type 1"
/db_xref="GeneID:94"
/db_xref="HGNC:HGNC:175"
/db_xref="MIM:601284"
exon 1..192
/gene="ACVRL1"
/gene_synonym="ACVRLK1; ALK-1; ALK1; HHT; HHT2; ORW2;
(...)GENBANK (continued)
(...)
ORIGIN
1 cccagtcccg ggaggctgcc gcgccagctg cgccgagcga gcccctcccc ggctccagcc
61 cggtccgggg ccgcgcccgg accccagccc gccgtccagc gctggcggtg caactgcggc
121 cgcgcggtgg aggggaggtg gccccggtcc gccgaaggct agcgccccgc cacccgcaga
181 gcgggcccag agggaccatg accttgggct cccccaggaa aggccttctg atgctgctga
241 tggccttggt gacccaggga gaccctgtga agccgtctcg gggcccgctg gtgacctgca
(...)
4081 aaattacact tctcgtacct ggagacgctg tttgtgggag cactgggctc atgcctggca
4141 cacaataggt ctgcaataaa ccatggttaa atcctga
//FASTA
>NM_000020.3 Homo sapiens activin A receptor like type 1 (ACVRL1), transcript variant 1, mRNA
CCCAGTCCCGGGAGGCTGCCGCGCCAGCTGCGCCGAGCGAGCCCCTCCCCGGCTCCAGCCCGGTCCGGGG
CCGCGCCCGGACCCCAGCCCGCCGTCCAGCGCTGGCGGTGCAACTGCGGCCGCGCGGTGGAGGGGAGGTG
GCCCCGGTCCGCCGAAGGCTAGCGCCCCGCCACCCGCAGAGCGGGCCCAGAGGGACCATGACCTTGGGCT
CCCCCAGGAAAGGCCTTCTGATGCTGCTGATGGCCTTGGTGACCCAGGGAGACCCTGTGAAGCCGTCTCG
GGGCCCGCTGGTGACCTGCACGTGTGAGAGCCCACATTGCAAGGGGCCTACCTGCCGGGGGGCCTGGTGC
ACAGTAGTGCTGGTGCGGGAGGAGGGGAGGCACCCCCAGGAACATCGGGGCTGCGGGAACTTGCACAGGG
AGCTCTGCAGGGGGCGCCCCACCGAGTTCGTCAACCACTACTGCTGCGACAGCCACCTCTGCAACCACAA
CGTGTCCCTGGTGCTGGAGGCCACCCAACCTCCTTCGGAGCAGCCGGGAACAGATGGCCAGCTGGCCCTG
ATCCTGGGCCCCGTGCTGGCCTTGCTGGCCCTGGTGGCCCTGGGTGTCCTGGGCCTGTGGCATGTCCGAC
(...)
GGCCCAATGGCCAGGGAGTGAAGGAGGTGGCGTTGCTGAGAGCAGTCTGCACATGCTTCTGTCTGAGTGC
AGGAAGGTGTTCCAGGGTCGAAATTACACTTCTCGTACCTGGAGACGCTGTTTGTGGGAGCACTGGGCTC
ATGCCTGGCACACAATAGGTCTGCAATAAACCATGGTTAAATCCTGAGFF/GTF/BED
- Interval formats
- Tab delimited
- Chromosomal coordinate, start, end, strand, and more
- BED Documentation
- GFF3 Documentation
BED
3 columns:
chr7 127471196 127472363
chr7 127472363 127473530
chr7 127473530 1274746976 columns:
chr1 134212701 134230065 Nuak2 8 +
chr1 134212701 134230065 Nuak2 7 +
chr1 33510655 33726603 Prim2, 14 -
chr1 25124320 25886552 Bai3, 31 -Bedtools
“Collectively, the bedtools utilities are a swiss-army knife of tools for a wide-range of genomics analysis tasks. The most widely-used tools enable genome arithmetic: that is, set theory on the genome. For example, bedtools allows one to intersect, merge, count, complement, and shuffle genomic intervals from multiple files in widely-used genomic file formats such as BAM, BED, GFF/GTF, VCF.”
.2bit
Installing twoBitToFa.
Downloading the mouse genome (assembly 9).
Bedtools (continued)
Given genes.bed:
chr1 134212701 134230065 Nuak2 8 +
chr1 134212701 134230065 Nuak2 7 +
chr1 33510655 33726603 Prim2 14 -
chr1 25124320 25886552 Bai3 31 -chr1 134210701 134212701 Nuak2 8 +
chr1 134210701 134212701 Nuak2 7 +
chr1 33726603 33728603 Prim2 14 -
chr1 25886552 25888552 Bai3 31 -promoters.fa
>chr1:134210701-134212701
TTCTGGCACTTGGTTGTTCT...GTTTTATAGCAATTCGGAAC
>chr1:134210701-134212701
TTCTGGCACTTGGTTGTTCT...GTTTTATAGCAATTCGGAAC
>chr1:33726603-33728603
TCTCCCAGTGGCGGGAGAGT...ATTTATTTTTATGTTTATAA
>chr1:25886552-25888552
TTGCGCCTTATCCAAGTGAA...TCCCAGGAACAAATCACCAGAutomation
- Let’s now create a script capturing all this information
Magic Line (Shebang)
- In a Unix-like operating system, the content of an executable is passed to the interpreter designated on the magic line.
- I am saving this to a file called
01_get_data.sh - Then, I make it executable
Test Your Assumptions
- You can test for the presence or absence of a file or a directory.
Temporary Space
- Sometimes you don’t want to create temporary files in your user account.
- These temporary files might be big and you don’t want them to be saved by the backup system, or your quota might not allow you to save them in your user space.
- Do not use
/tmp/, this is temporary storage for the operating system, and sometimes the partition is rather small. - Use
/var/tmp/or a designated space, such as/scratch.- Beware! The system will automatically remove those files after a given period of time.
Script (continued)
#! /bin/bash
# Sample Bash script to download a genome and extract information
INPUT=genes.bed
if [ ! -f $INPUT ]; then
echo "file not found: $INPUT"
exit 1
fi
PROJECT=csi5180-demo
# Process ID and time stamp as suffix
TMP_DIR=/var/tmp/$PROJECT-`date +"%FT%H%M%S"`-$$
if [ -d TMP_DIR ]; then
echo "$TMP_DIR exists!"
exit 1
fiScript (continued)
# Creating the temporary directory
mkdir $TMP_DIR
# The URL where the mouse genome version 9 (MM9) can be found
MM9_URL=http://hgdownload.cse.ucsc.edu/goldenpath/mm9/bigZips/mm9.2bit
# Where to save the mouse genome as a fasta file
MM9_FILE_NAME=$TMP_DIR/mm9.fa
# Download an uncompress the genome
twoBitToFa -udcDir=$TMP_DIR $MM9_URL stdout > $MM9_FILE_NAME
# URL of the file containing the size of each chromosome
MM9_SIZE_URL=http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/mm9.chrom.sizes
MM9_SIZE_FILE_NAME=$TMP_DIR/mm9.chromsizes
# Downloading the size file (to the current directory)
curl $MM9_SIZE_URL > $MM9_SIZE_FILE_NAMEScript (continued)
REST
Interactions Using HTTP
Representational State Transfer (REST)
- Client and server interactions using HTTP (hypertext transfer protocol)
- Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47, W636–W641 (2019).
- Tarkowska, A. et al. Eleven quick tips to build a usable REST API for life sciences. PLoS Comput Biol 14, e1006542 (2018).
Documentation
Examples:
ENSEMBL: GET sequence/id/:id
ENSEMBL: GET sequence/id/:id
>ENST00000288602.11
CCGCTCGGGCCCCGGCTCTCGGTTATAAGATGGCGGCGCTGAGCGGTGGCGGTGGTGGCG
GCGCGGAGCCGGGCCAGGCTCTGTTCAACGGGGACATGGAGCCCGAGGCCGGCGCCGGCG
CCGGCGCCGCGGCCTCTTCGGCTGCGGACCCTGCCATTCCGGAGGAGGTGTGGAATATCA
AACAAATGATTAAGTTGACACAGGAACATATAGAGGCCCTATTGGACAAATTTGGTGGGG
AGCATAATCCACCATCAATATATCTGGAGGCCTATGAAGAATACACCAGCAAGCTAGATG
CACTCCAACAAAGAGAACAACAGTTATTGGAATCTCTGGGGAACGGAACTGATTTTTCTG
TTTCTAGCTCTGCATCAATGGATACCGTTACATCTTCTTCCTCTTCTAGCCTTTCAGTGC
TACCTTCATCTCTTTCAGTTTTTCAAAATCCCACAGATGTGGCACGGAGCAACCCCAAGT
CACCACAAAAACCTATCGTTAGAGTCTTCCTGCCCAACAAACAGAGGACAGTGGTACCTG
CAAGGTGTGGAGTTACAGTCCGAGACAGTCTAAAGAAAGCACTGATGATGAGAGGTCTAA
TCCCAGAGTGCTGTGCTGTTTACAGAATTCAGGATGGAGAGAAGAAACCAATTGGTTGGG
ACACTGATATTTCCTGGCTTACTGGAGAAGAATTGCATGTGGAAGTGTTGGAGAATGTTC
CACTTACAACACACAACTTTGTACGAAAAACGTTTTTCACCTTAGCATTTTGTGACTTTT
GTCGAAAGCTGCTTTTCCAGGGTTTCCGCTGTCAAACATGTGGTTATAAATTTCACCAGC
GTTGTAGTACAGAAGTTCCACTGATGTGTGTTAATTATGACCAACTTGATTTGCTGTTTG
TCTCCAAGTTCTTTGAACACCACCCAATACCACAGGAAGAGGCGTCCTTAGCAGAGACTG
CCCTAACATCTGGATCATCCCCTTCCGCACCCGCCTCGGACTCTATTGGGCCCCAAATTC
TCACCAGTCCGTCTCCTTCAAAATCCATTCCAATTCCACAGCCCTTCCGACCAGCAGATG
AAGATCATCGAAATCAATTTGGGCAACGAGACCGATCCTCATCAGCTCCCAATGTGCATA
TAAACACAATAGAACCTGTCAATATTGATGACTTGATTAGAGACCAAGGATTTCGTGGTG
ATGGAGCCCCTTTGAACCAGCTGATGCGCTGTCTTCGGAAATACCAATCCCGGACTCCCA
GTCCCCTCCTACATTCTGTCCCCAGTGAAATAGTGTTTGATTTTGAGCCTGGCCCAGTGT
TCAGAGGATCAACCACAGGTTTGTCTGCTACCCCCCCTGCCTCATTACCTGGCTCACTAA
CTAACGTGAAAGCCTTACAGAAATCTCCAGGACCTCAGCGAGAAAGGAAGTCATCTTCAT
CCTCAGAAGACAGGAATCGAATGAAAACACTTGGTAGACGGGACTCGAGTGATGATTGGG
AGATTCCTGATGGGCAGATTACAGTGGGACAAAGAATTGGATCTGGATCATTTGGAACAG
TCTACAAGGGAAAGTGGCATGGTGATGTGGCAGTGAAAATGTTGAATGTGACAGCACCTA
CACCTCAGCAGTTACAAGCCTTCAAAAATGAAGTAGGAGTACTCAGGAAAACACGACATG
TGAATATCCTACTCTTCATGGGCTATTCCACAAAGCCACAACTGGCTATTGTTACCCAGT
GGTGTGAGGGCTCCAGCTTGTATCACCATCTCCATATCATTGAGACCAAATTTGAGATGA
TCAAACTTATAGATATTGCACGACAGACTGCACAGGGCATGGATTACTTACACGCCAAGT
CAATCATCCACAGAGACCTCAAGAGTAATAATATATTTCTTCATGAAGACCTCACAGTAA
AAATAGGTGATTTTGGTCTAGCTACAGTGAAATCTCGATGGAGTGGGTCCCATCAGTTTG
AACAGTTGTCTGGATCCATTTTGTGGATGGCACCAGAAGTCATCAGAATGCAAGATAAAA
ATCCATACAGCTTTCAGTCAGATGTATATGCATTTGGAATTGTTCTGTATGAATTGATGA
CTGGACAGTTACCTTATTCAAACATCAACAACAGGGACCAGATAATTTTTATGGTGGGAC
GAGGATACCTGTCTCCAGATCTCAGTAAGGTACGGAGTAACTGTCCAAAAGCCATGAAGA
GATTAATGGCAGAGTGCCTCAAAAAGAAAAGAGATGAGAGACCACTCTTTCCCCAAATTC
TCGCCTCTATTGAGCTGCTGGCCCGCTCATTGCCAAAAATTCACCGCAGTGCATCAGAAC
CCTCCTTGAATCGGGCTGGTTTCCAAACAGAGGATTTTAGTCTATATGCTTGTGCTTCTC
CAAAAACACCCATCCAGGCAGGGGGATATGGTGCGTTTCCTGTCCACTGAAACAAATGAG
TGAGAGAGTTCAGGAGAGTAGCAACAAAAGGAAAATAAATGAACATATGTTTGCTTATAT
GTTAAATTGAATAAAATACTCTCTTTTTTTTTAAGGTGAAC
Making a Python Script Executable
A Python script can also be made executable.
ENCODE
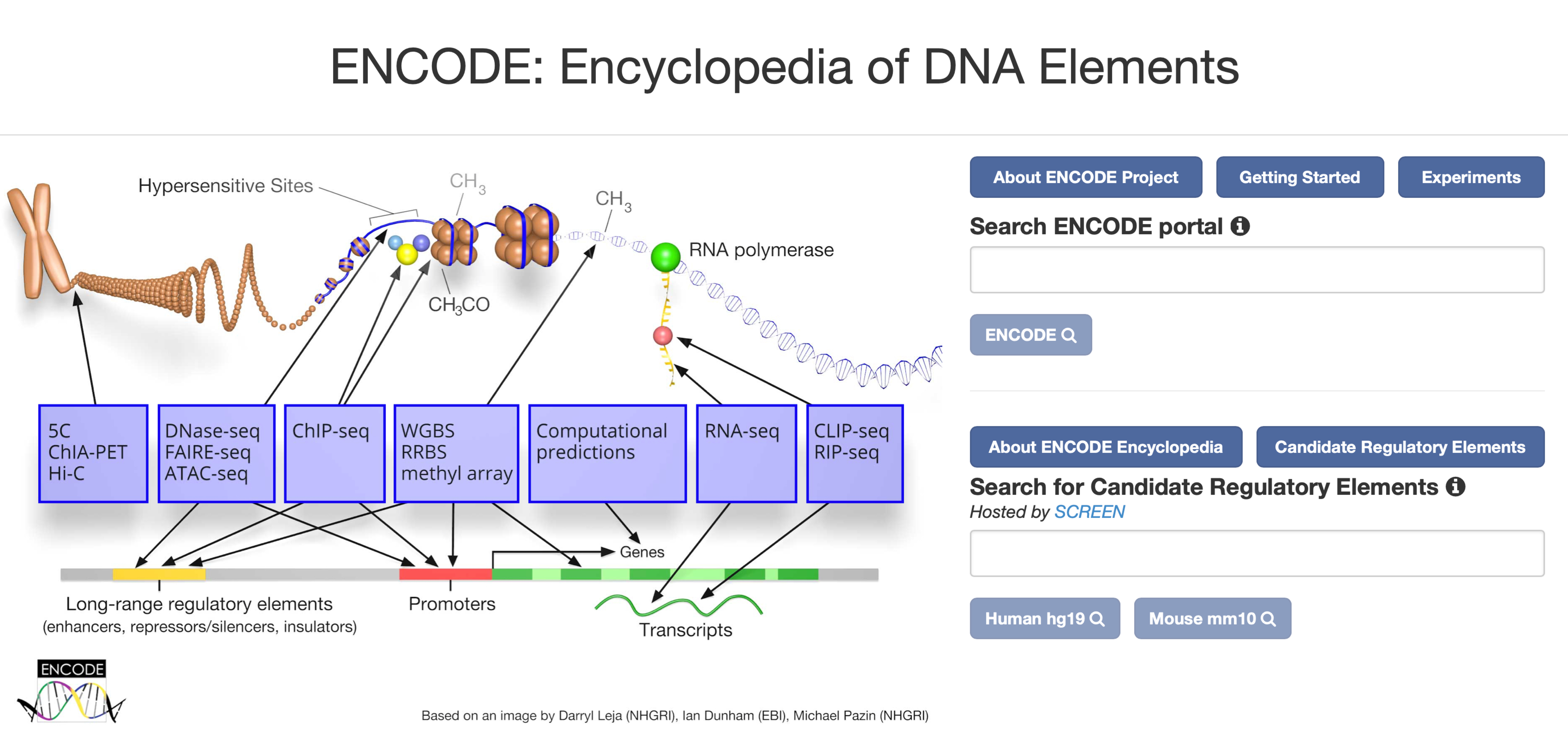
Pipelines
Discussion Groups
Tutorials
Guidelines
File Structure
- Bioinformatics projects typically produce a substantial volume of files. Employing a standardized directory structure across all projects significantly enhances the efficiency of information retrieval.
- Utilizing standardized tools, such as cookie-cutter templates, encourages uniformity in experimental protocols both within a single laboratory and across different institutions.
Robust Research (Vince Buffalo)
- Pay attention to your experimental design
- Write code for humans, write code for computers
- Let the computer do the work
- Write down your assumptions and test them (unit testing)
- Use existing libraries
- Treat data as read-only
Reproducible Research (Vince Buffalo)
- Share your source code and your data
- Meta-data:
- Versions of the software and databases you are using
- Write down the parameters or better yet, make it a script
- One README file per directory
- Make figures, statistics, and tables from scripts
- Not only is this more scientific, it is almost certain that you will need to redo your analyses!
Other Considerations
- Consider using a (distributed) version control system
- Git/GitHub has become the de facto standard
- Features
- Manage changes in your documents
- In a distributed version control system, each developer has its own version of the source code
- Multiple contributors
- Creating/merging multiple branches
- https://git-scm.com/doc
Prologue
Summary
- Strive to make your research robust and reproducible
- UNIX is the preferred environment for bioinformatics and machine learning
- Conda/Anaconda/Bioconda will simplify your life tremendously
- NCBI/EBI/DDBJ are the major repositories for bioinformatics data
- There are many specialized bioinformatics repositories
- GenBank, Fasta, and BED are examples of file formats
- Entrez Direct/REST
- Pipelines
Resources
Bioinformatics Data Skills

- Vince Buffalo. Bioinformatics Data Skills: Reproducible and Robust Research with Open Source Tools. O’Reilly Media, 2015.
A Practical Introduction to…

- Röbbe Wünschiers. Computational Biology - A Practical Introduction to BioData Processing and Analysis with Linux, MySQL, and R. Springer, 2013.
- link.springer.com/book/10.1007/978-3-642-34749-8
A Practical Introduction to…

- Röbbe Wünschiers. Computational Biology - A Practical Introduction to Bio Data Juggling with Worked Examples. Springer, 2025.
- link.springer.com/book/9783031703133
- Source Code
- Available March 2, 2025
The Biostar Handbook

- The Biostar Handbook: Bioinformatics data analysis guide, 2024
- www.biostarhandbook.com
- biostar.myshopify.com (student edition = $37)
Ten (10) simple rules for…
- Sandve, G. K., Nekrutenko, A., Taylor, J. & Hovig, E. Ten Simple Rules for Reproducible Computational Research. PLoS Comput Biol 9, (2013).
- Boulesteix, A.-L. Ten simple rules for reducing overoptimistic reporting in methodological computational research. PLoS Comput Biol 11, e1004191 (2015).
- Prlic, A. & Procter, J. B. Ten Simple Rules for the Open Development of Scientific Software. PLoS Comput Biol 8, e1002802 (2012).
Ten (10) simple rules for…
- Perez-Riverol, Y. et al. Ten Simple Rules for Taking Advantage of Git and GitHub. PLoS Comput Biol 12, e1004947 (2016).
- Sholler, D. et al. Ten simple rules for helping newcomers become contributors to open projects. PLoS Comput Biol 15, e1007296 (2019).
- Rule, A. et al. Ten simple rules for writing and sharing computational analyses in Jupyter Notebooks. PLoS Comput Biol 15, e1007007 (2019).
Ten (10) simple rules for…
- Osborne, J. M. et al. Ten simple rules for effective computational research. PLoS Comput Biol 10, e1003506 (2014).
- Elofsson, A. et al. Ten simple rules on how to create open access and reproducible molecular simulations of biological systems. PLoS Comput Biol 15, e1006649 (2019).
- Lee, B. D. Ten simple rules for documenting scientific software. PLoS Comput Biol 14, e1006561 (2018).
Ten (10) simple rules for…
- Carey, M. A. & Papin, J. A. Ten simple rules for biologists learning to program. PLoS Comput Biol 14, e1005871 (2018).
- Zook, M. et al. Ten simple rules for responsible big data research. PLoS Comput Biol 13, e1005399 (2017).
Next Lecture
- Fundamentals of Machine Learning
References
Marcel Turcotte
School of Electrical Engineering and Computer Science (EECS)
University of Ottawa